When working with sequencing datasets of ecological interest, an interesting problem is how to tease out the genetic diversity present in the population being sequenced. Usually, assembly software simply aligns the short read sequences, and determines the consensus sequence based on the majority vote of each position. However, we may wish to seperately assemble each haplotype (version) of each gene.
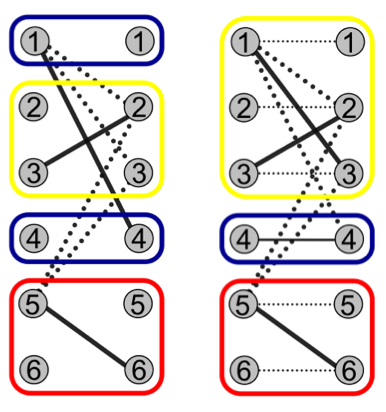
We formulate this as a graph problem, where short reads that overlap are considered nodes in a graph that share an edge if they should go in different haplotypes. We then need to minimally “color” the graph (assign a minimum number of colors to nodes such that connected nodes always get different colors, upper right). This normally NP-Hard problem is solvable in cubic time given the linear input data.

Further, the software we developed (Hapler) additionally maximizes the number of versions that only have a single read in them (by slightly modifying the construction), isolating sequencing errors as rare erroneous haplotypes. Paper.
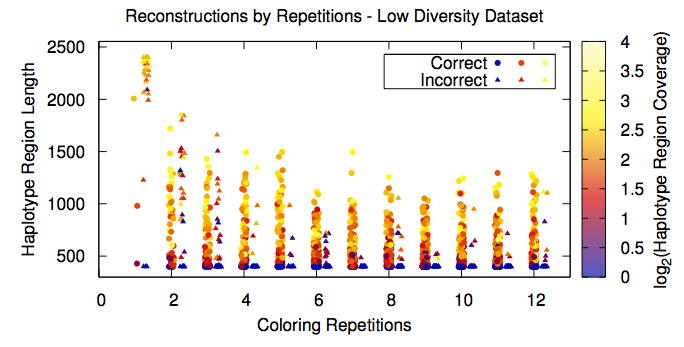
Given the model described above, there are many possible solutions representing a parsimonious haplotype assembly, most of which represtent erroneous chimeric assemblies. Psuedo-randomly sampling from the space of haplotypings (taking advantage of the ordered nature of the coloring algorithm and rearranging the input matrix), we can then keep only the commonalities between solutions. While this process reduces the size of the haplotype assemblies, correctness is dramatically increased and returned solutions are much more likely to represent biological reality. Paper.